
1. Angew:PCN-601氧化还原活性位用于甲醇氧化和CO2还原

华南师范大学刘江和兰亚乾等人建立复合CO2 电还原(HCER)的配位催化剂模型体系(具有活性氧化位点的Ni8-TET、具有活性还原位点的Ni-TPP和具有氧化还原活性位点的PCN-601)。发现PCN-601能完成甲醇的阳极氧化反应以及阴极CO2还原反应,FEHCOOH和FECO都超过90%。并且通过光照可以进一步提高性能(FE接近100%)。

DFT计算当对体系施加电场时,卟啉中心的 Ni 带相对的负电荷,而 Ni8 簇中的 Ni 带相对的正电荷。意味着只要对系统施加电场,电荷就会从 Ni8 簇转移到卟啉;紧接着计算 Ni8 簇和 Ni-TPP 上 CO2RR 和 MOR 的自由能图。Ni8 簇的 CO2RR 活性比 Ni-TPP 差(ΔG0max = 1.50 vs. 0.68 eV,ΔG0max代表所有基本步骤中的最大标准吉布斯能量),而 Ni8 簇的 MOR 活性优于 Ni-TPP Ni-TPP (ΔG0max = -0.64 vs -0.55 eV)。
Ni-TPP 上 CO2RR 的速率决定步骤 (RDS) 是 CO2+H2O→COOH+OH–,考虑到Ni卟啉将接收来自 Ni8 簇的电子,当在Ni卟啉中加入一个电子发现RDS的反应能降低0.26 eV,表明电荷转移有利于CO2RR。
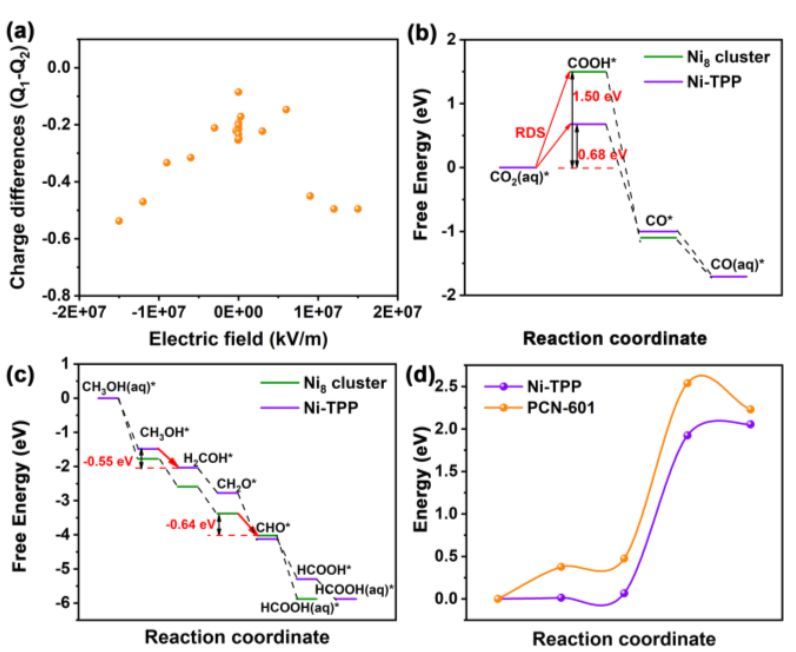
此外,NEB模拟CO2+H+e→COOH*过程发现吡唑和苯具有不同的亲氧性,与苯环相比,吡唑基团的活化能显着降低,证明PCN-601的吡唑基团可以降低DRS过程能垒。Sheng-Nan Sun, Long-Zhang Dong et al. Redox-Active Crystalline Coordination Catalyst for Hybrid Electrocatalytic Methanol Oxidation and CO2 Reduction. Angew. Chem. Int. Ed. 2022
2. CJC: ZnN4P/C 活性位催化剂用于ORR

重庆大学Tayyaba Najam,彭立山和魏子栋等人报道P、N双掺杂碳骨架负载的单原子Zn催化剂(Zn-N4P/C)用于氧还原反应(ORR);Zn-N4P/C 催化剂表现出和Pt/C相当的 ORR 活性(E1/2 = 0.86 V)和优于 Pt/C的稳定性;在Zn-air电池测试中中的最大峰值功率密度为249.6 mW cm-2,比电容为779 mAh g-1,以及150 小时的充放电循环稳定性。
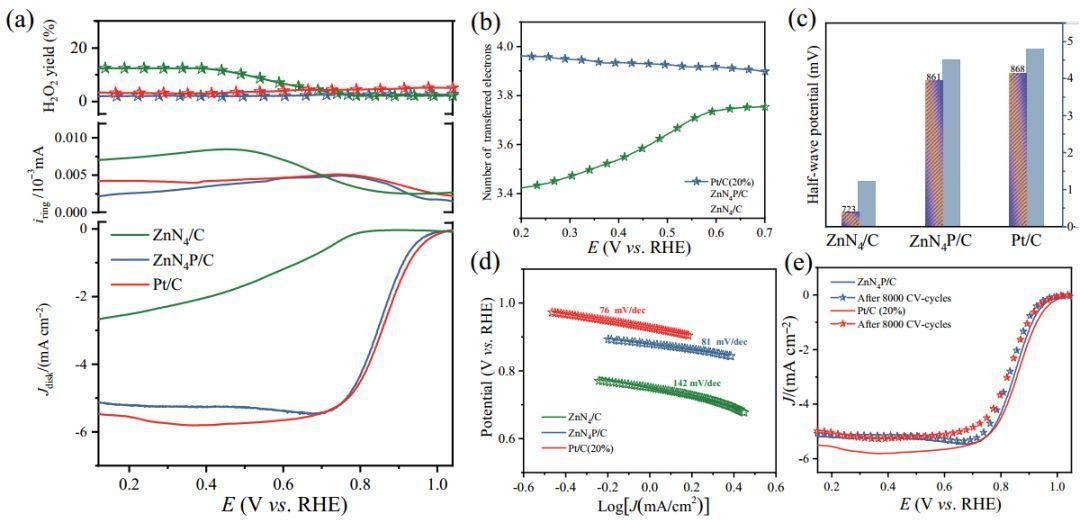
以ZnN4/C和 ZnN4P/C为结构模型进行DFT理论计算。发现P 掺杂的 Zn-N4 部分 (Zn-N4-P) 显示 Zn 原子周围的电子密度比基面负载的 Zn-N4 位点更高,表明ZnN4P/C 上的电荷密度重新分布;研究ZnN4/C 和 ZnN4P/C 的部分态密度 (PDOS)发现ZnN4P/C 中 Zn 的 d 带隙的负位移比 ZnN4/C 中的 Zn 原子稍小,表明通过 P 掺杂获得 Zn-N4 活性位显示增强的局部电子电导率。
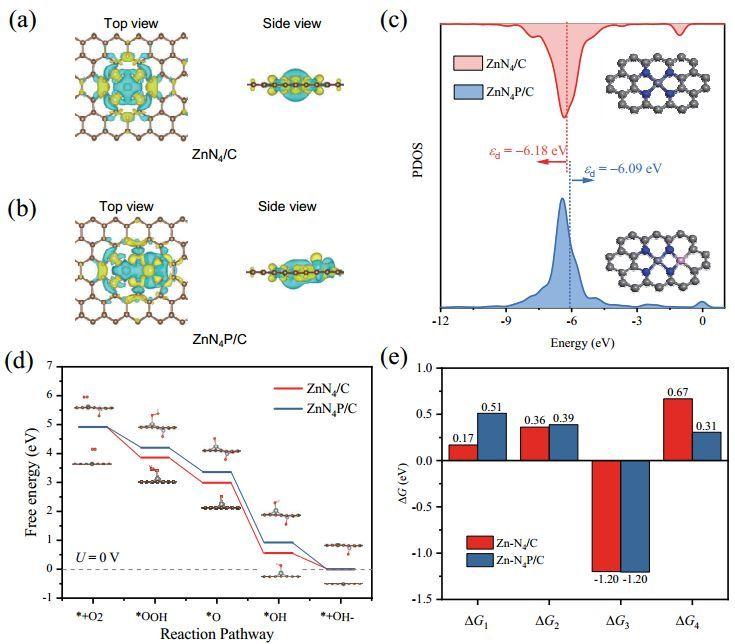
计算吉布斯自由能图确认最后的电子转移步骤(*OH 解吸)是速率决定步骤(RDS),表明 Zn-O 亲和力的减弱可以促进ORR活性。因此,进一步研究ZnN4/C 和 ZnN4P/C 的吸附和脱附性能;与 Zn-N4 相比,ZnN4P/C 的 ΔGOH*(*OH 的吸附自由能)值大得多,表明对氧分子的结合能力较低,OH*脱附过程更快;*OH 在 Zn-N4/C 中的强吸附导致RDS步骤的能垒更大,而 ZnN4P/C 样品可以更好地调节*OH 结合强度,最终降低RDS能垒。
Syed Shoaib Ahmad Shah, Tayyaba Najam et al. Modulating the microenvironment structure of single Zn atom: ZnN4P/C active site for boosted oxygen reduction reaction. Chinese J. Catal. 2022. 8, 2193-2201.
3. Angew: 用于电催化质子还原的聚乙二醇Sn卟啉配合物

辛辛那提大学Jianbing “Jimmy” Jiang和田纳西大学Konstantinos D. Vogiatzis等人验证基于主族元素的Sn卟啉配合物,作为质子还原的有效分子电催化剂的适用性。聚乙二醇Sn卟啉复合物 (SnPEGP) 在乙腈(MeCN)电解液中以及三氟乙酸 (TFA) 质子源中显示出高催化活性(-1.7 V vs.Fc/Fc+, 电流密度为-4.6 mA/cm2)和高选择性(-1.7 V vs.Fc/Fc+, H2 法拉第效率为 94% );H2产生的最大周转频率 (TOFmax) 为 1099 s-1。

DFT计算用于深入研究SnPEGP催化质子还原机理。根据化学步骤(C,质子化)和电子转移(E)步骤的顺序,HER 的双电子/双质子转移步骤可以通过多种途径发生:CCEE、CECE、CEEC、EECC、ECEC 和 ECCE。Sn(II) 物种是通过以金属为中心的两电子还原以及 2 个 Cl– 轴向配体的损失形成。反应的第一步要么涉及系统的还原,要么涉及 Sn 或 N 原子的质子化。
研究发现还原的 SnPEGP 催化剂(中间体 1)的自由能差(ΔG298)为 0.95 eV(施加过电势为 0.05),其能量低于任何质子化配合物(0.20 – 0.66 eV)。因此,初始还原步骤是有利的;在此之后,与第二次还原相比,最有可能发生质子化。第二次还原具有 2.08 eV 的自由能差(施加过电势为 1.18 eV),高于任何质子化配合物。
Sn 原子(中间体 2)的质子化是唯一的放热步骤(-0.13 eV),而 N 原子的质子化是吸热过程。在形成中间体2之后,SnPEGP 表面上发生质子化或还原。任何一个可用 N 原子的第二次质子化以约 0.90 eV 的自由能差进行,而第二次还原(中间体 3)以 1.26 eV 的自由能差进行,随着施加的过电位降低至 0.36 eV。
因此,反应的第三步很可能涉及还原,表明SnPEGP 催化剂上的 HER 是通过ECEC 途径进行,即还原之后是质子化,然后重复。反应的最后一步涉及以相似的自由能差进行的任何一个 N 原子的质子化。在最接近 PEG 部分的 N 原子上发生质子化的中间体(中间体 4A)显示出 0.16 eV 的自由能差,而另一个 N 原子(4B)的质子化以 0.34 eV 的自由能差进行。由于这种小的能量差异,最终的质子化步骤可能发生在任一N原子。

两质子/两电子转移步骤之后,研究导致 H2 形成的键解离步骤相关的动力学。断裂 N-H 和 Sn-H 键形成 H2 的动力学势垒为 0.67 eV (15.4 kcal/mol)(4A)以及0.82 eV (18.9 kcal/mol)(4B)。HER 最有可能在中间体 4A 形成后继续进行;最后发现 H2 与 SnPEGP 催化剂的弱结合(0.13 eV)有利于催化剂再生。
Ashwin Chaturvedi, Gavin A. McCarver et al. A PEGylated Tin-Porphyrin Complex for Electrocatalytic Proton Reduction: Mechanistic Insights into Main-Group Element Catalysis. Angew. Chem. Int. Ed. 2022, e202206325
4. AM:无定形NiB2用于高效电合成H2O2

苏州大学康振辉教授,刘阳教授,陈子亮副教授,柏林工业大学Prashanth W. Menezes等人通过用不同量的 BH4– 定向还原 Ni2+,灵活调节无定形NiB2纳米结构的内在活性,以实现高效的 2e-ORR;无定形 NiB2 在 0.4 V电压下,2e– 选择性接近99%,在较宽的电位范围内选择性超过 93% ;将NiB2组装到气体扩散电极后,实现 4.753 mol gcat-1 h-1 的超高 H2O2 产率。
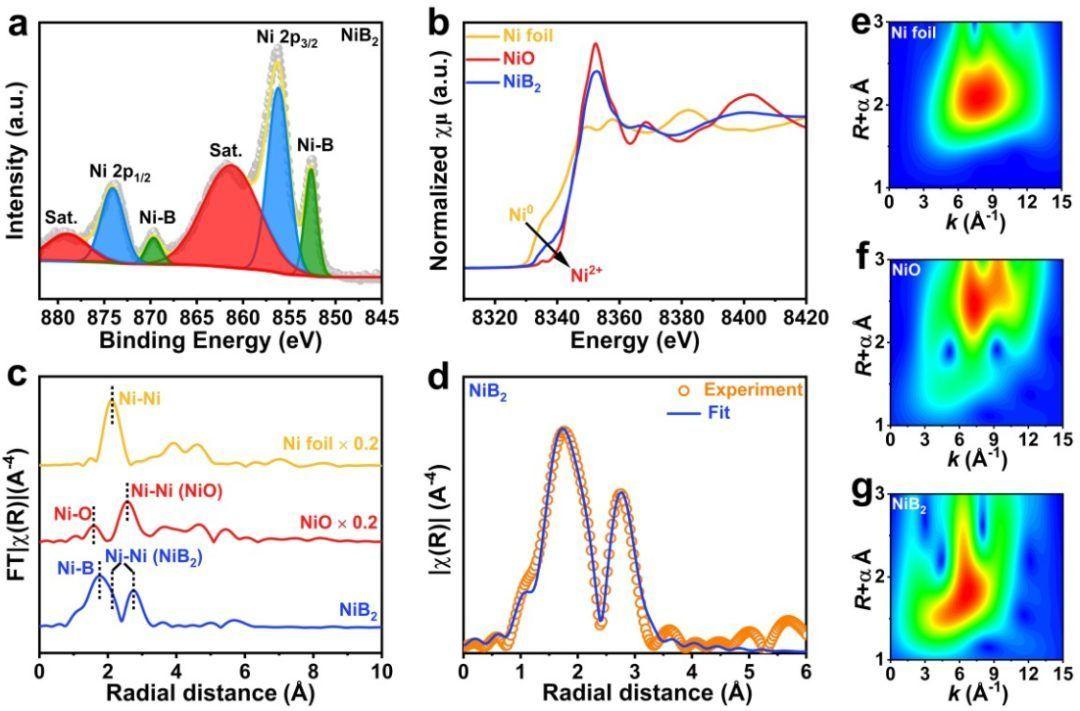
DFT计算硼化物对中间体 *OOH (ΔGOOH*) 的吸附自由能,发现NiB2 的 ΔGOOH* 值 (3.75 eV) 最接近理想值 3.52 eV,表明对 *OOH 的吸附和解吸能力达到最佳平衡,有利于 O-O 键的保留和 H2O2 的产生;使用 ΔGOOH*描绘2e-ORR 路径的极限电位-火山图,发现NiB2最接近火山图顶端,表明产生H2O2的活性最高;*OOH 转化为 *O 的自由吸附能垒约为 +0.21 V,大于 0.7 V *OOH 转化为 *O 的自由吸附能垒(-0.23 eV),表明高的二电子还原选择性;当 Ni 与 B 的比例较高时,例如 Ni3B、Ni2B 和 NiB,*OOH 的吸附模型倾向于侧向模式,不利于O-O键保留,当 B 与 Ni 的比例较高时,B 原子可以有效地隔离相邻 Ni 原子,导致*OOH 的端吸附模式,并有利于 O-O 键的保存;研究底物和*OOH之间的差分电荷密度分布发现NiB2在底物和 *OOH 之间表现出适度的电荷相互作用,从而提高催化活性。以上结果表明,Ni电子结构和吸附模型可以通过 Ni 和 B 之间的原子比进行调整,从而优化 ΔGOOH*。

Jie Wu, Meilin Hou, et al. Composition Engineering of Amorphous Nickel Boride Nanoarchitectures Enabling Highly Efficient Electrosynthesis of Hydrogen Peroxide. Adv. Mater. 2022
5. ACS Catal.:MOF负载的双铜位点用于CO2还原制C2+ 产物

中山大学廖培钦等人证明金属有机骨架 Cu-HITP (HITP = 2,3,6,7,10,11-六亚氨基苯并苯) 晶体表面上的双铜位点非常适合催化*CO 与*COH 偶联形成*OCCOH,在 KHCO3 电解液中实现C2+ 还原产物的高选择性,C2+产物的法拉第效率为 75(3)%,C2H4的法拉第效率为 51(1)%。

DFT计算结果表明,速率限制步骤是*CO转化为*COH,反应能为0.95 eV;关键中间体*OCCOH的形成能仅为-0.74 eV,低的C-C耦合反应能表明通过*OCCOH中间体很容易在Cu-HITP表面形成C-C键;另外,两个相邻 *CO 物种之间的距离为 2.72 Å,因此*CO物种在 Cu-HITP 表面上直接形成 *OCCO 中间体是热力学不利的;中间体 *CO 和 *CHO 之间的距离为 3.25 Å,*CO 与 *COH 偶联生成 *OCCOH 是形成 C-C 键的最热力学主要途径。
进一步研究Cu(100)和Cu(111)表面上*CO和*COH物种之间的C-C耦合。在Cu-HITP表面,*CO和*COH物质耦合形成CC键的反应能为-0.74 eV,低于Cu(100)和Cu(111)晶面的反应能;此外,对于 *CO + *COH → *OCCOH,Cu-HITP 催化的自由能垒与 Cu(111) 和 Cu(100) 表面上的自由能垒相比最低,表明与 Cu(100) 和 Cu(111) 晶面相比,Cu-HITP 中活性位点之间的距离 (3.4 Å) 更适合 *CO 与 *COH 耦合形成 *OCCOH 的低能途径。
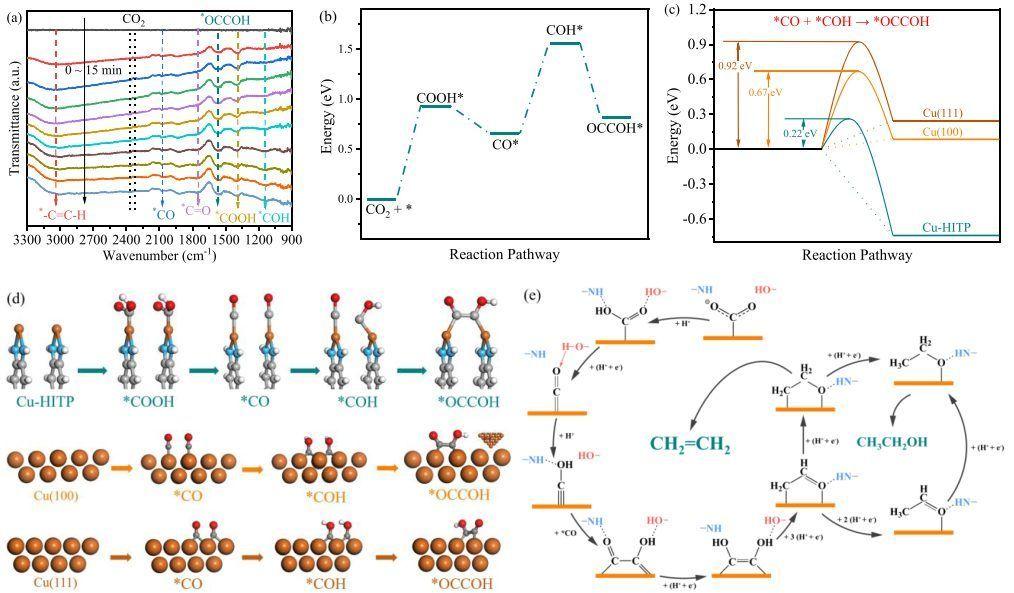
Zhen-Hua Zhao, Hao-Lin Zhu, et al. Polydopamine Coating of a Metal−Organic Framework with BiCopper Sites for Highly Selective Electroreduction of CO2 to C2+ Products. ACS Catal. 2022, 12, 7986−7993
6. AFM:吡咯氮稳定的单价Ni单原子电催化剂用于CO2还原

韩国全北大学Do Hwan Kim和延世大学Do Hwan Kim等人开发软模板辅助技术来合成吡咯 N4-Ni 位点以及不同的 N 型缺陷,以协同提高 CO2RR 性能。最佳催化剂在 -0.6 V 的低电位下实现 94% 的 CO 法拉第效率,在 -1 V 下实现 59.6 mA cm-2 的 CO 部分电流密度。

DFT计算表明石墨 N 和吡啶 N 上 COOH* 中间体形成的自由能分别为 2.57 和 0.30 eV,远高于吡咯 N(-0.40 eV),意味着吡咯 N 上的有利的COOH* 动力学;在 CO* 解吸过程中,由于 CO* 的强结合,吡咯 N 位点必须克服更高的脱附自由能。这些结果表明,N 掺杂位点对 CO2RR 的催化性能受到不利的COOH* 形成或 CO* 缓慢脱附的限制。
此外,吡咯 N 物种附近的 C 呈现 COOH* (-0.23 eV) 和 CO* (0.23 eV) 形成的最小自由能变化,表明平衡的吸附和脱附过程增强CO2RR性能。具有孤对电子的吡咯 N 增加了相邻碳 (δ–) 上的电子密度,这导致吡咯 N-C 键长 (1.338 Å) 比吡啶 N-C 键长 (1.349 Å) 更短,吡咯 N-C 位点处电子密度的增加有利于 CO2 的吸附,从而加速 CO2RR过程。
此外,晶体轨道汉密尔顿分布(COHP) 分析表明,吡咯 N 附近的 C 与 COOH* 的 C 具有更强的键合相互作用。吡咯C-C的键合强度为-8.12 eV,高于吡啶N-C(-5.42 eV),表明吡咯 C 和 COOH* 中间体的 C 之间的相互作用更强。因此,吡咯 C 位点降低了 COOH* 形成的自由能,提高 CO2RR 活性。

Ramireddy Boppella, Muthu Austeria P et al. Pyrrolic N-Stabilized Monovalent Ni Single-Atom Electrocatalyst for Efcient CO2 Reduction: Identifying the Role of Pyrrolic–N and Synergistic Electrocatalysis. Adv. Funct. Mater. 2022, 2202351
7. Angew. :Rh金属烯上原子分散 MoOx 促进碱性HER

吉林大学范锦昌和崔小强等人报告使用一锅溶剂热法合成锚定在 Rh 金属烯上的原子分散的 MoOx 物种,催化剂结构最大程度地暴露氧化物-金属界面,产生具有超高碱性 HER 活性的 MoOx-Rh 催化剂,在 50 mV 的过电位下获得了 2.32 A mgRh-1 的质量活性,是商业 Pt/C 的 11.8 倍。
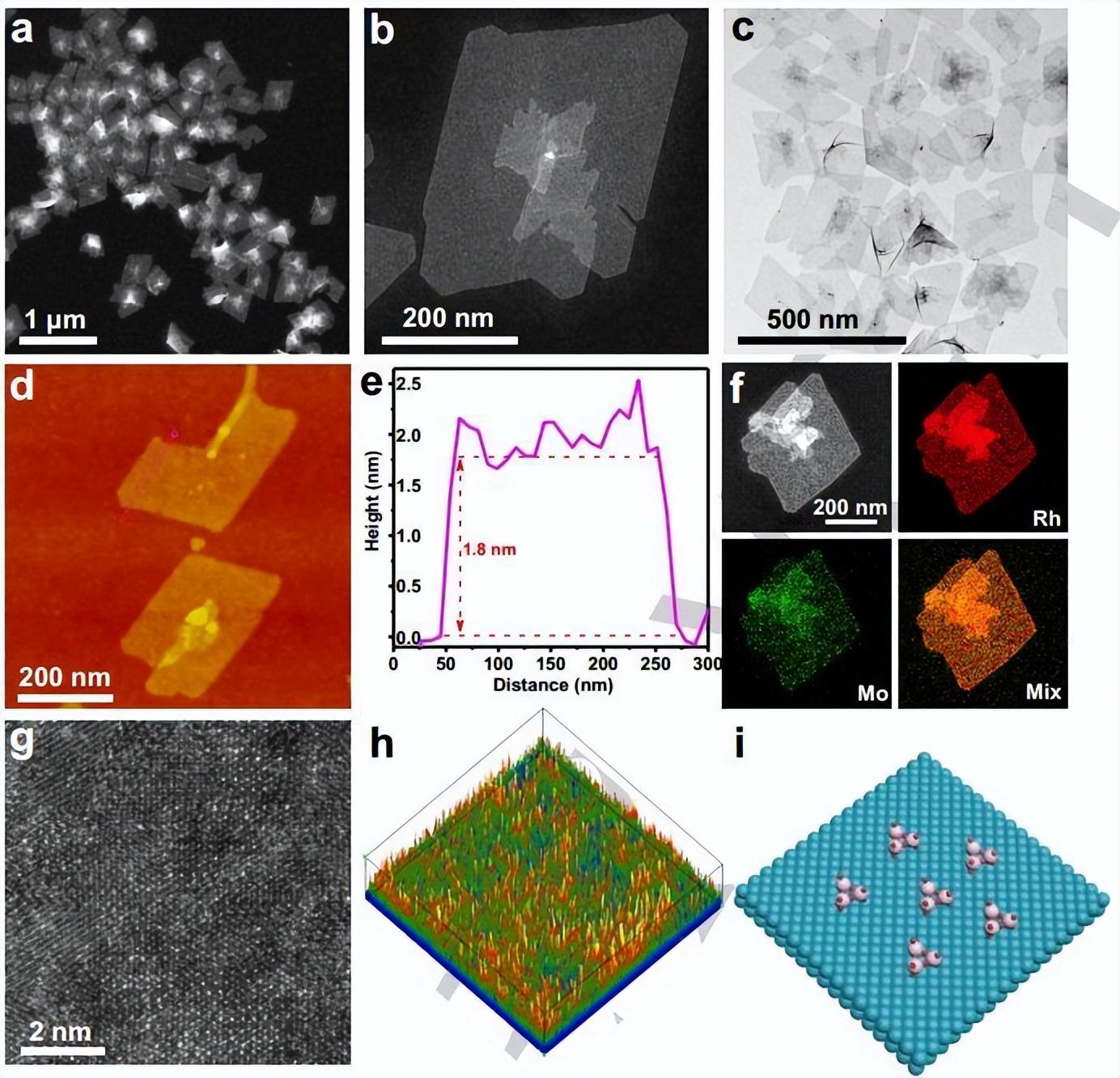
DFT计算阐述MoOx-Rh 中MoOx 和 Rh 在碱性 HER过程中的协同作用。研究发现MoOx 簇锚定在 Rh(111) 表面, 在 MoOx-Rh 金属烯界面有丰富的电子积累;DFT计算表明原子分散的MoOx物种通过O原子锚定在Rh金属烯的表面,强的共价结合能为(-3.32 eV)使得MoOx物种在HER过程中具有高稳定性。通常,缓慢的 H2O 解离过程限制了碱性 HER 的反应动力学,因此进一步研究H2O在催化剂表面的吸附和解离。
与Rh位点相比,界面处的不饱和致密MoOx位点优先吸附和解离H2O分子,而随后的H*吸附和H2解吸发生在相邻的Rh原子上;特别是,H2O 在 MoOx-Rh 上的吸附能为 -0.51 eV,比 Pt (111) 表面 (0.05 eV) 或 Rh (111) 表面 (-0.08 eV) 的吸附能强;MoOx-Rh 对 H2O 的更强吸附有利于碱性 HER过程,即促进 Volmer 反应。此外,与 Pt(111) 表面 (0.55 eV) 和 Rh (111) 表面 (-0.09 eV) 相比,MoOx-Rh 显示出 -0.40 eV 的优异 H2O 解离活性。

DFT进一步计算H* 的吸附自由能 (ΔGH*)。Rh(111) 表面上的 ΔGH* 值很大,表明氢原子在 Rh 催化剂上强结合,不利于H2的脱附;相比之下,对于MoOx-Rh 金属烯,当覆盖率超过 0.77 单层时,H* 将倾向于吸附在界面 Rh 位点上,并且 ΔGH* 值接近最佳值(ΔGH*= 0),表明 MoOx 和 Rh 之间的界面有利于 H2O 解离、H* 吸附和 H2 解吸。
Jiandong Wu, Jinchang Fan et al. Atomically Dispersed MoOx on Rhodium Metallene Boosts Electrocatalyzed Alkaline Hydrogen Evolution. Angew. Chem. Int. Ed. 2022, e202207512
8. Nat. Commu.:Co@CoO核壳催化剂催化氢解5-羟甲基糠醛制2,5-二甲基呋喃

华东理工大学龚学庆、王艳芹联合曼彻斯特大学杨四海等人报道独特的Co@CoO核壳结构催化剂,其将生物质衍生的 5-羟甲基糠醛高效氢解为 2,5-二甲基呋喃;并且研究发现催化活性位点位于具有氧空位的 CoO 壳上,而不是金属 Co。
DFT计算阐明CoO 壳上氧空位对于HMF 的氢解的重要性。首先计算H2 在 CoO(100) 和 CoO(100)-Ov 表面上的吸附和解离的能量分布。H2在CoO(100)和CoO(100)-Ov处的吸附能分别为0.25和1.63 eV,表明CoO(100)-Ov具有更强的结合能力;重要的是, CoO(100) 上H2异裂过程吸热 0.54 eV,势垒为0.60 eV,而在 CoO(100)-OV 表面上,吸热仅为0.36 eV,并且具有较低的势垒(0.56 eV);因此,CoO(100)-OV 有利于 H2 的异裂。
计算 H2 在 CoO(100)-OV 表面上的均裂发现OV可以辅助H2在催化剂表面裂解为两个Hδ−,并且H2的均裂过程放热(0.23 eV),能垒为 0.51 eV;表明CoO(100)-OV 表面上H2均裂比异裂更容易,并且两个Hδ-的形成可能使CoO(100)-OV表面比CoO更活跃。

进一步计算CoO(100)-OV表面HMF的吸附、活化及转化成DMF的完整反应路径。研究发现,HMF 首先吸附在 CoO(100)-OV 表面,吸附放热高达 2.22 eV,高于 H2 的吸附放热(1.63 eV);此外,HMF通过-CH=O的O原子垂直吸附在CoO(100)-Ov表面,填补材料的氧空位;随后,H2吸附在 CoO(100)-Ov 表面,并放热 0.75 eV;吸附的 H2 通过异裂过程以 0.60 eV 的势垒被活化,其中一个 H 原子与 Co 原子键合,而另一个被 O 原子占据;Co-Hδ-的电负性H首先攻击HMF中吸附的-CH=O的正电Cδ+,该过程放热 0.63 eV,需要克服 0.63 eV 的势垒;Hδ+攻击-CH2~O的O原子是速率决定步骤,该过程吸热0.97 eV,势垒为1.42 eV;然后,在 BHMF 氢解过程中生成 HMMF和 H2O,分别需要克服 0.84 eV 和 1.3 eV 的能垒;在 HMMF 氢解生成 DMF 和 H2O 的过程中,需要克服 1.23 和 0.33 eV 的能垒。
Xiang, S., Dong, L., Wang, ZQ. et al. A unique Co@CoO catalyst for hydrogenolysis of biomass-derived 5-hydroxymethylfurfural to 2,5-dimethylfuran. Nat Commun 13, 3657 (2022)
———END———
限 时 特 惠: 本站每日持续更新海量各大内部创业教程,永久会员只需98元,全站资源免费下载 点击查看详情
站 长 微 信: yjxmw518



